Analyze the data
used:
The DNA methylation and gene expression data of human
BRCA tissues from the publicly available
The Cancer Genome Atlas database.
Choose the 109-th batch Illumina Infinium HumanMethylation450 Beadchip and RNASeqV2
level
three data. The human
reference genome annotation file RefSeq gene (hg19) from UCSC.
Based
on the Illumina Infinium HM450
array annotation file (hg19), the genome was divided into the following six
regions:
TSS1500
, 5' UTR
, 3' UTR
, TSS200
, gene body
, 1stexon
Genome-specific sites:
CGI
DHS
enhancer
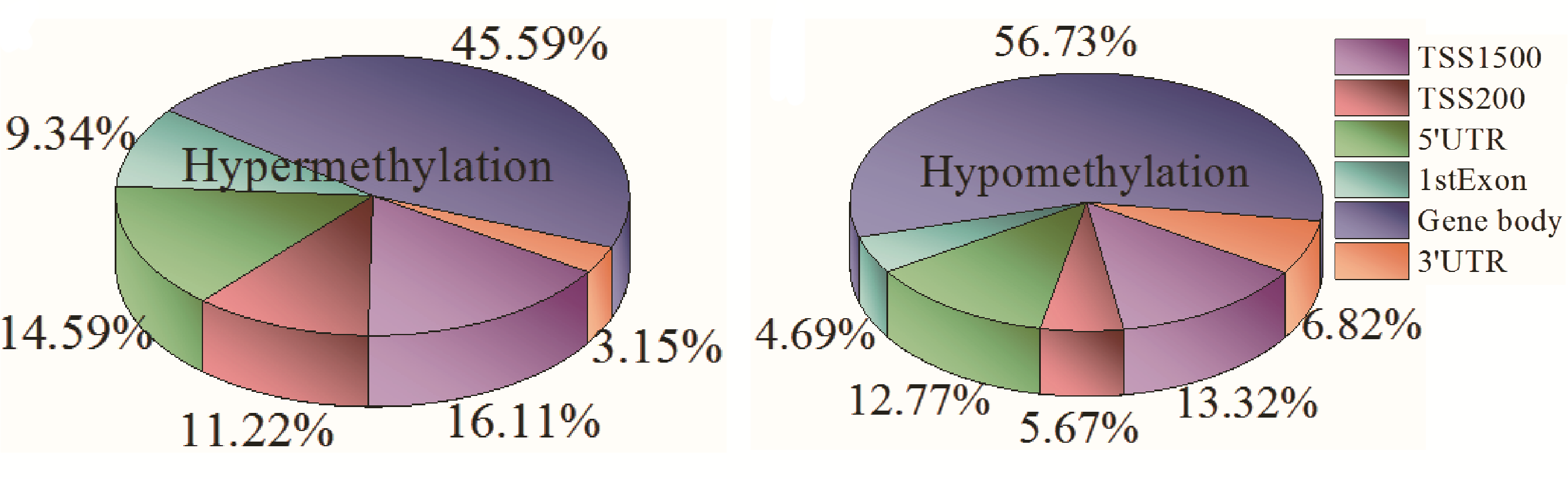
Analysis result of
figure
above:
For the distributions of hypermethylated
CpG sites in the six regions, the proportion of gene body
region is the highest, which reaches at 45.59% of the total
number of hypermethylation sites of the six regions. However, TSS1500, TSS200, 5'UTR,
1stExon, and 3'UTR account for 16.11%, 11.22%, 14.59%, 9.34%, and 3.15%,
respectively. The distributions of hypomethylated CpG sites
of the six regions are that gene body region is also the
highest proportion, 56.73% of the total number of hypomethylation sites of the six
regions. Second, TSS1500 is
13.32%. Nevertheless, the proportion of 1stExon in hypomethylation is the lowest, 4.69%.
Then, TSS200 is 5.67%.
5'UTR is 12.77%, and 3'UTR is 6.82%. Gene body region
enriches the highest of hyper- and hypomethylation sites
compared with the other five regions in breast tumor tissue.
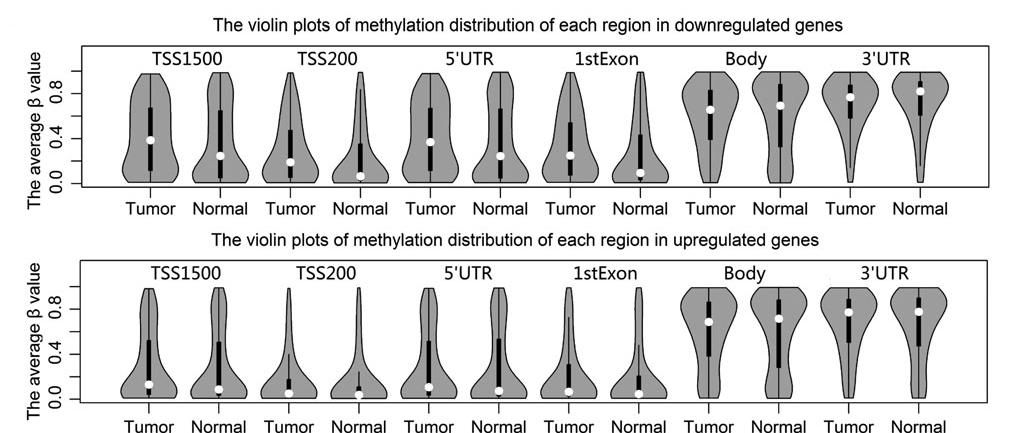
Analysis result of
figure
above:
The methylation distribution difference between the two tissues in each region is
more visible in downregulated genes than upregulated genes,
particularly promoter region (including TSS1500, TSS200,
5'UTR, and 1stExon regions). DNA methylation has a very
strong effect on the downregulation of gene expression in
breast cancer, especially promoter methylation. The average b values in TSS1500, TSS200,
5'UTR, and
1stExon regions are lower, whereas gene body region and
3'UTR are higher for down- and upregulated genes in breast
cancer tissue. In the downregulated genes, the
methylation distributions of the two tissues are markedly
different at TSS1500, TSS200, 5'UTR, and 1stExon regions.
For instance, the distributions of TSS1500 region and 5'UTR
are more dispersed, their density of high methylation is greater
in tumor than normal tissue, and their methylation
medians in tumor are higher than that of normal tissue.

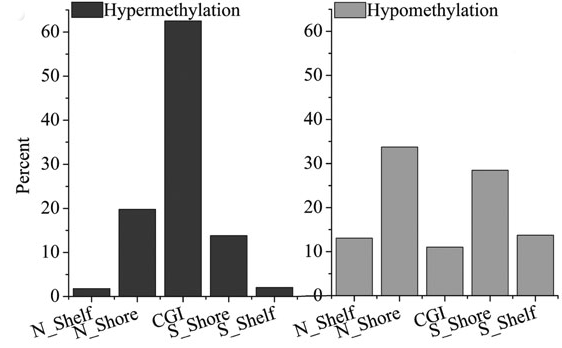
Analysis result of
figure
above:
Hypermethylation sites,
44.54%, enrich in CGIs and 31.42% and 24.04% of these
sites locate on DHS and enhancer regions, respectively. For
the hypomethylation sites, only 14.01% of them distribute in
CGIs, and 22.84% of them are DHSs, whereas the number of
these sites enriched in enhancer region is up to 63.15%. In
the downregulated genes, hypermethylation sites mainly
distribute in CGIs. In the upregulated genes, hypomethylation sites principally
distribute in enhancer regions. It is illustrated that the downregulation of genes is
related to CGI
hypermethylation, and the upregulation of genes is closely
correlated with enhancer hypomethylation.
Analysis result of
figure
above:
In these regions, the hypermethylation sites in downregulated genes are primarily
concentrated in CGI and its proportion is the highest. In contrast, the hypomethylation
proportion in CGI for up-regulated
genes is the lowest. About 62% of the hypomethylation sites in
upregulated genes are enriched in N_Shore and S_Shore regions. In other words, the
hypomethylation of shore region
plays important roles in gene expression regulation for breast
carcinoma.
If you want to know more, please click here.
More detailed
information can be found in the "Detail" button on the search page.